RELIWE - Predicting Biomolecular Interactions
by Bernd Kramer, Christian Lemmen, Thomas Lengauer and Matthias Rarey
The biological activity of pharmaceutical compounds is often based on controlling certain reactions in complex metabolic processes of the human organism. Therefore developing small organic molecules (ligands) that strongly bind to a special protein molecule (receptor) is an important problem. The binding process is also called molecular docking. The forces dominating the docking process are weak short-range interactions that require the molecules to fit both geometrically and chemically. To predict the strength of binding and the three-dimensional structure of potential ligands in the complex of ligand and receptor is one of the central questions in the development of new drugs (rational drug design).
The RELIWE project aims at developing efficient algorithms and data structures for the docking and superposition problems and their implementation in a software tool.
Docking problem
The docking problem asks for the geometry (binding mode) as well as for the strength of binding of a ligand to a structurally known receptor. If a reliable prediction is possible, the affinity to the same receptor of many chemical com-pounds that are available in databases can be tested rapidly, saving energy and time expended on laboratory work.
Superposition problem
For many applications in the pharma-ceutical industry, the 3D structure of the receptor is unknown because it cannot be determined routinely by experiment. Thus, often we know only a set of ligands together with their measured biological activities towards the receptor. Statistical methods are used to extract the relevant geometric and chemical features of the ligands. From this 'fingerprint' of specific interactions one can draw conclusions about the structure of the receptor at the binding site and the potential activity of alternative ligands. A central aspect in this context is the relative orientation of the ligands in 3D space. This is again a typical task for the computer, which can predict plausible relative orientations.
Approach to the docking problem
Beginning with the three-dimensional structure of the receptor, our docking algorithm identifies potential interaction partners inside the binding-site and represents them using appropriate interaction geometries. By modeling chemical aspects of the docking problem directly, we avoid generating solutions which are meaningful only geometrically but not chemically. In order to handle ligand flexibility, a small user-defined part of the ligand, called the base fragment, is placed first. This process uses algorithms that are adapted from computer vision. Subsequently, the ligand is re-constructed incrementally inside the binding-site.
Efficient algorithms and data structures for three-dimensional range queries, for the storage of partial placements, and for evaluating the energy function have been developed, in order to reduce the time and space requirements. Essential parts of the model and of the algorithmic engines can be modified to suit the solution of the superposition problem.
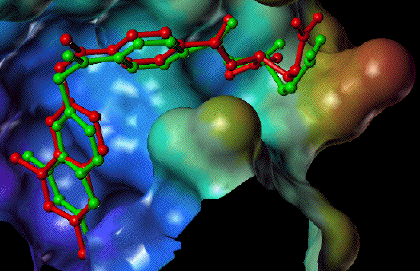
Comparison of the predicted placement of a ligand to the experimentally observed complex.Status of the project
A prototype of our docking tool FlexX has been in operation since the beginning of 1995. The program is already in use by our industrial partners for first field tests. On a set of important benchmarks, the program has been able to reproduce the experimentally observed receptor-ligand complexes within the experimental accuracy, in a few minutes on a workstation.
RELIWE on the web: http://www.gmd.de/SCAI/alg/reliwe/reliwe_home.html
Please contact:
Bernd Kramer - GMD
Tel: +49 2241 14 2276
E-mail: bernd.kramer@gmd.de
Christian Lemmen - GMD
Tel: +49 2241 14 2481
E-mail: christian.lemmen@gmd.de
Thomas Lengauer - GMD
Tel: +49 2241 14 2777
E-mail: thomas.lengauer@gmd.de
Matthias Rarey - GMD
Tel: +49 2241 14 2476
E-mail: matthias.rarey@gmd.de